


The pharmaceutical and medical device industries are some of the most heavily regulated sectors globally and for a good reasonProducts manufactured in these industries impact people’s health and well-being directly. The big Regulatory organizations including but not limited to the FDA (Food and Drug Administration) in the U.S., the EMA (European Medicines Agency), and the ISO (International Organization for Standardization), TGA (Therapeutic Goods Administration), MHRA (Medicines and Healthcare products Regulatory Agency), CDSCO (Central Drug Standard Control Organisation), ANVISA (Agência Nacional de VigilânciaSanitária), WHO all require companies to demonstrate that their facilities meet rigorous operational standards.
Failure to comply with these standards can have severe consequences. These include:
CQV helps prevent these issues by ensuring that your equipment and processes are in perfect working order, from day one of operation.
Commissioning, Qualification & Validation (CQV) is an integral part of the life sciences and pharmaceutical sector. It is a very detail-oriented process that requires the right mix of knowledge, experience and diligence to correctly place Facilities/Equipment/Utilities into use.
The main difference between Commissioning and Q&V are the guidelines followed. Commissioning is an exercise made from a Good Engineering Practice (GEP) point of view. Q&V is an exercise made from a Good Manufacturing Practice (GMP) point of view.
The objective of CQV process is to demonstrate that a system was designed, installed and performs as per design requirements and/or for its intended use:
The International Society for Pharmaceutical Engineering (ISPE) has provided over the years several guidelines on how Qualification & Validation activities should be implemented.
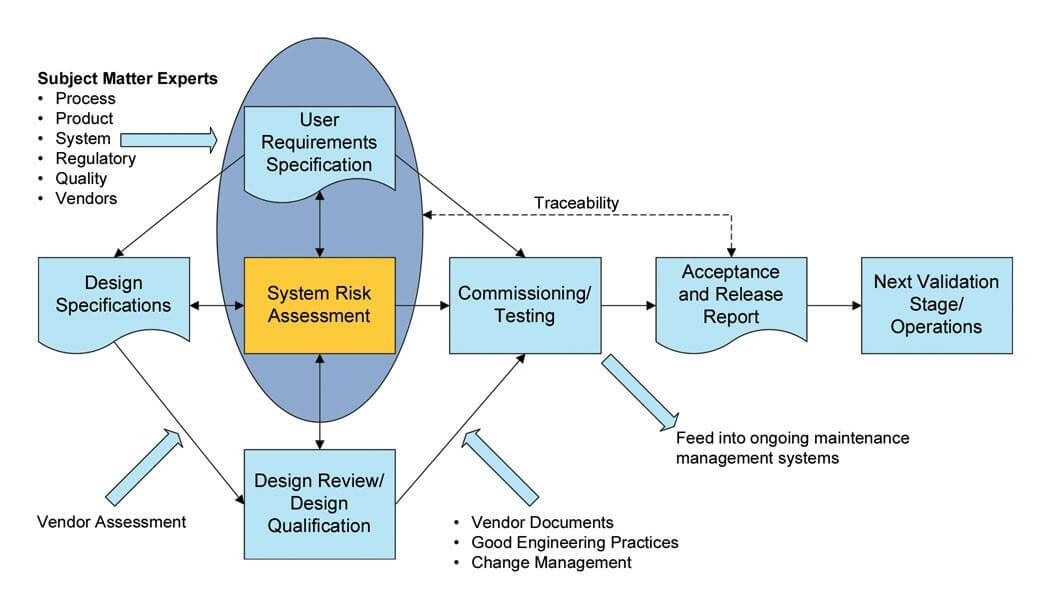
Within their guidelines, one of the most widely used Lifecycle Development models is the “V” Model, which is a framework or structure for undertaking the design, execution, commissioning and qualification of a design project. It was first promoted by the International Society of Pharmaceutical Engineers (ISPE) back in 1994 in the first edition of their Good Automated Manufacturing Practices guideline (GAMP).
The “V” Model creates a solid basis for testing the functionality of the system against the original design specifications and proving that the delivered technical solution achieves the desired outcome.

Qualification and Validation activities are mandatory to comply with GMP, Commissioning activities are not. However, a robust commissioning strategy will ensure your facilities, equipment and utilities were installed adequately and are operating according to the specified conditions.
The User Requirements Specification is a document that can be and should be used to its’ full potential by procurement, engineering, quality, operations (i.e., lean improvements) and validation departments instead of “just another document to complete and file away”.
A GMP Impact Assessment is a crucial step for the CQV activities because it distinguishes between Direct, Indirect and No Impact Systems, as well as Critical and Non-critical Components optimizing the Commissioning and Qualification & Validation effort.
CQV activities are not a one time only activity. Periodic review / requalification routines must be implemented to revise the system status, associated change controls and deviations or operational control results with the objective of detecting any trends and implement preventive measures avoiding future deviations.

|
303-304, Sahyog Elina, VIP Main Road |
|
|
510-11, Wing-A, 73 East Avenue, |
|
| +91 9879594538 |
 GAMP Services Limited 2026. All Rights Reserved.
GAMP Services Limited 2026. All Rights Reserved.